moleditpy_pyscf-calculator
MoleditPy PySCF Calculator Plugin
A powerful, user-friendly GUI interface for performing quantum chemistry calculations using PySCF. This plugin provides an intuitive workflow for configuring calculations, managing jobs, and visualizing molecular electronic structure.
Research-Grade Power with Educational Clarity: Transform abstract quantum mechanics into tangible, interactive discoveries. Built on the industrial-strength PySCF engine, this plugin delivers rigorous accuracy for researchers while offering an intuitive visual interface that makes it an indispensable platform for mastering Physical Chemistry or Organic Chemistry. Whether you are a researcher performing rapid conformational scans and transition state searches to screen candidates, or a student decoding the principles of molecular orbital theory, this tool bridges the gap between complex algorithms and chemical insight. From predicting reactivity with HOMO/LUMO visualizations to mapping detailed Potential Energy Surfaces, it empowers users at all levels to visualize, analyze, and understand the fundamental forces driving chemical change.
Tutorial
Master quantum chemistry calculations in MoleditPy with step-by-step interactive guides. These tutorials demonstrate how to integrate structural modeling with electronic structure theory using PySCF.
Gallery
Features
Calculation Capabilities
- Job Types: Single Point Energy, Geometry Optimization, Frequency Analysis, Transition State Optimization, Rigid & Relaxed Surface Scans.
- Methods: RHF, UHF, RKS, UKS (DFT).
- Functionals: Support for standard functionals (B3LYP, PBE, etc.) via PySCF.
- Advanced Configuration: Control over Basis Sets, Charge/Spin, Symmetry, Max Cycles, Convergence Tolerance, CPU Threads, and Memory.
- Settings Management: Manually save your preferred configuration (Method, Basis, Resources) as the default for future sessions.
Visualization & Analysis
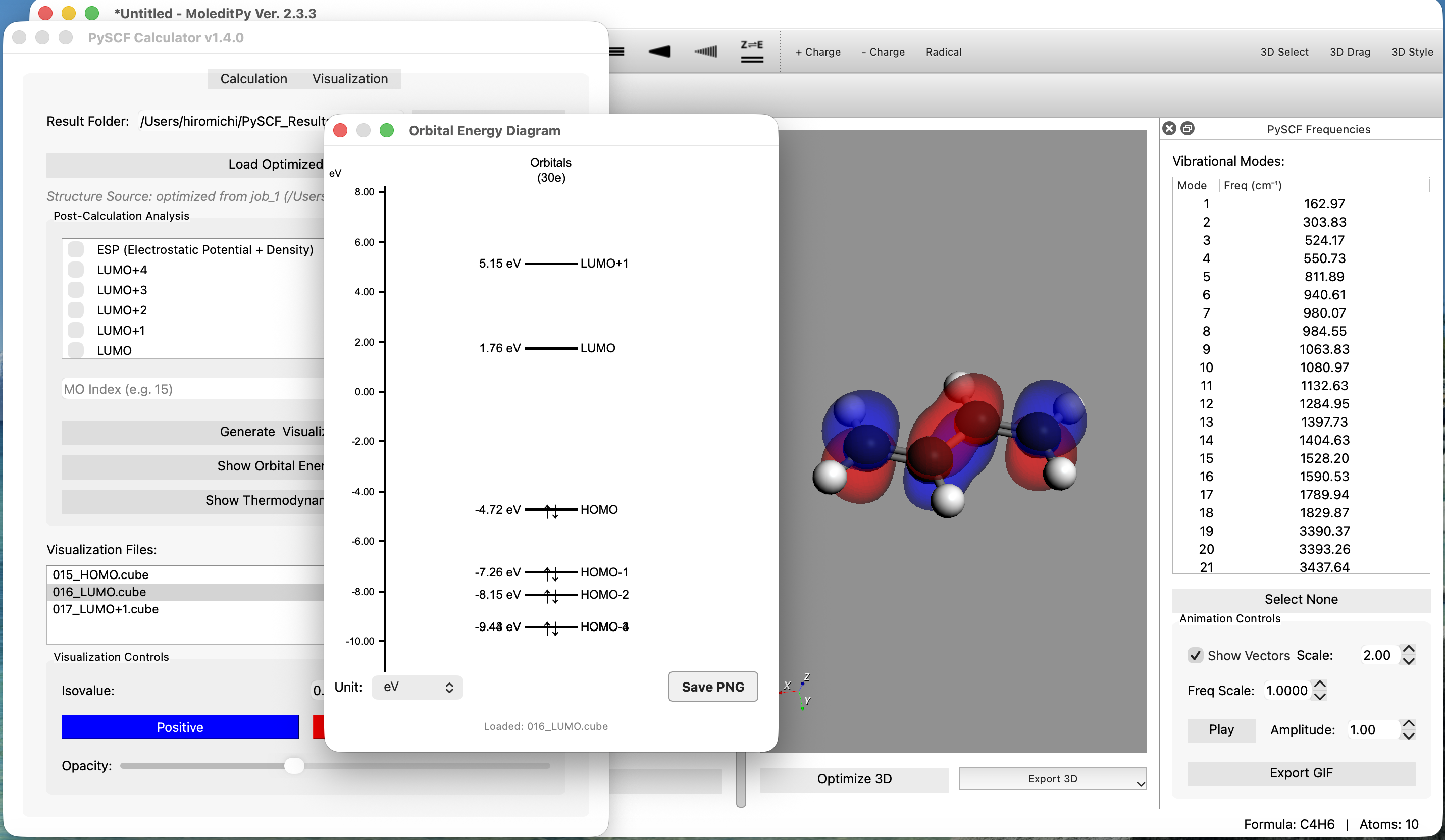
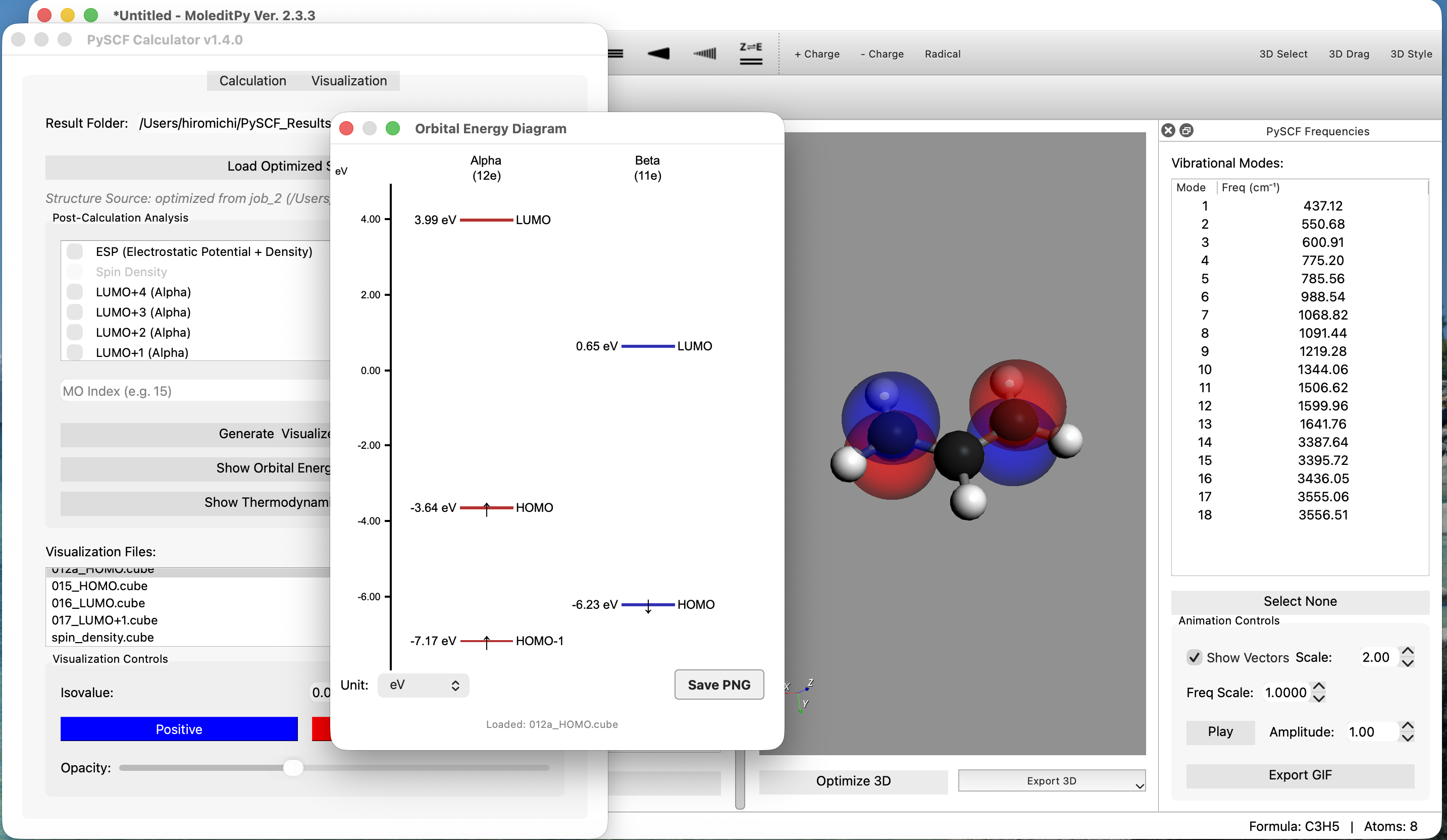
- Interactive Orbital Energy Diagram:
- Automatically displays HOMO/LUMO energies and gaps.
- Interactive Loading: Click on any orbital line (e.g., HOMO) to automatically load and visualize its Electron Density (Cube file).
- On-Demand Generation: If a cube file is missing, clicking the orbital prompts you to generate it instantly without re-running the full job.
- Navigation:
- Zoom: Drag up/down to zoom in/out of energy levels.
- Pan: Scroll (Touchpad compatible) to move the energy view up/down.
- Reset: Double-click to restore the default view formatted to the HOMO-LUMO gap.
- Clean UI:
- Selectable units (eV / Hartree) with nice, round axis ticks.
- “Save to PNG” feature that automatically hides UI controls for publication-quality figures.
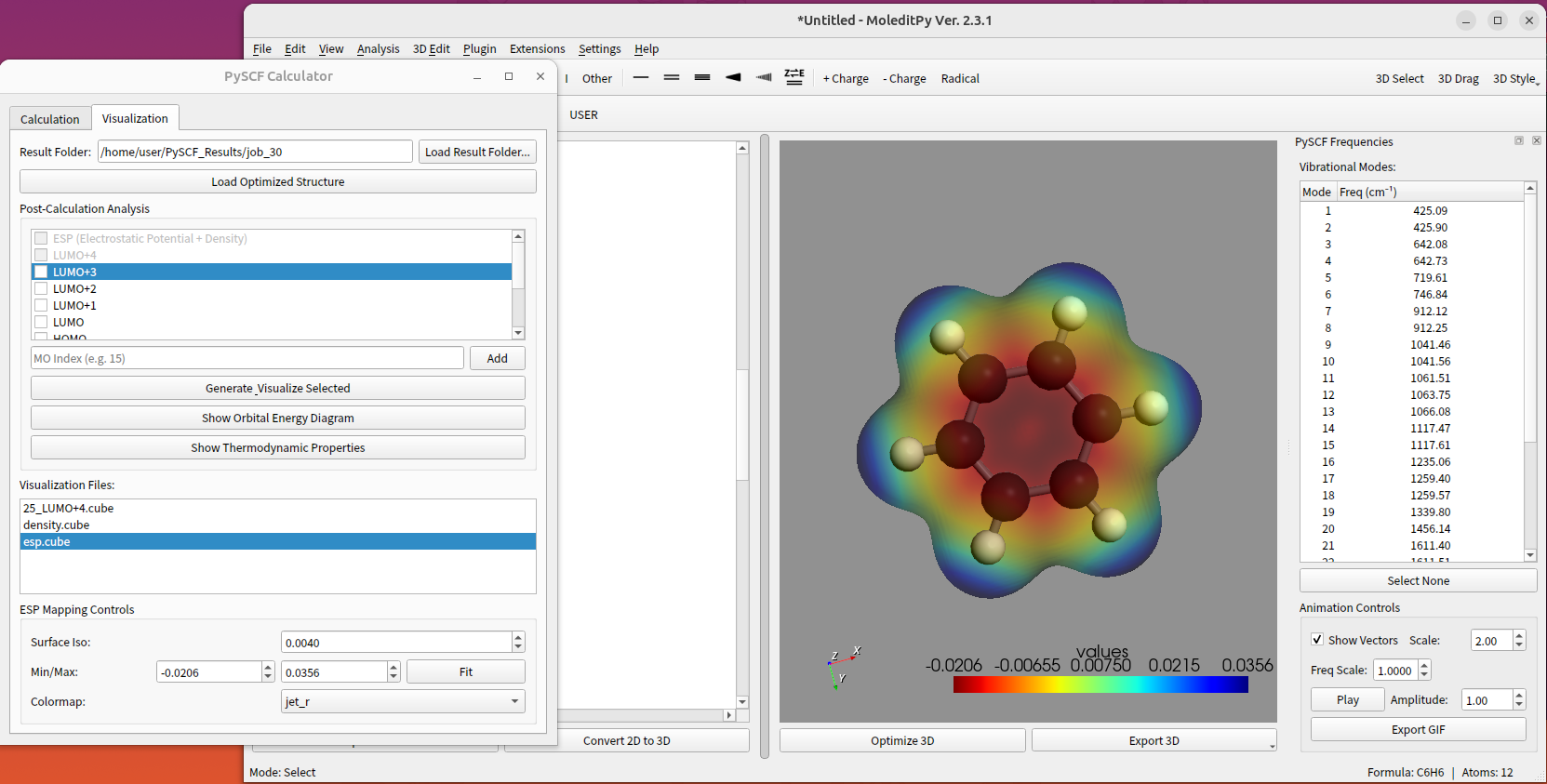
- Property Analysis:
- Generate standard .cube files for Molecular Orbitals (MOs).
- Compute and visualize Electron Density, Spin Density, and Electrostatic Potential (ESP).
- Handles Open-Shell (UHF) density correctly.
- Thermodynamic Properties: Calculate and view Enthalpy, Entropy, Gibbs Free Energy, and ZPE in a structured table format.
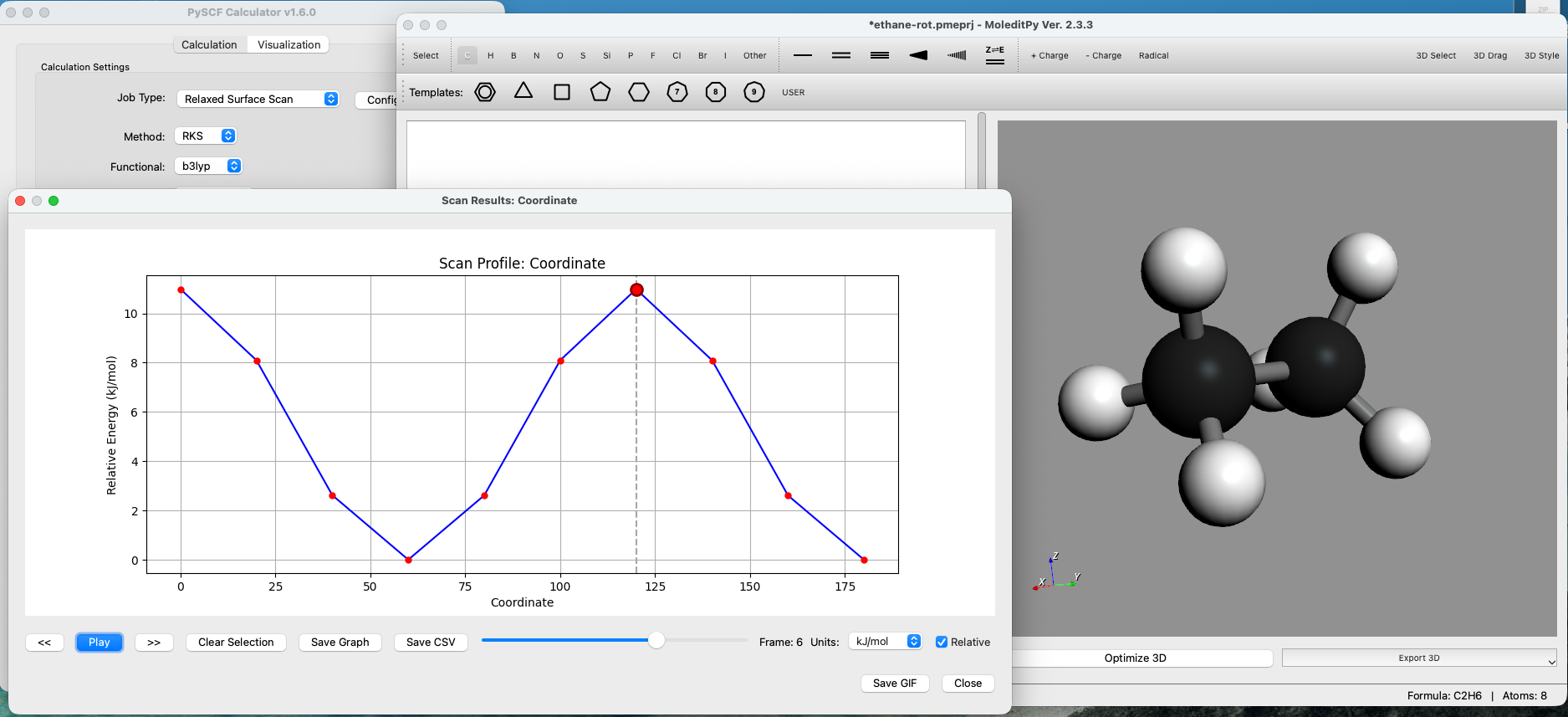
- Surface Scans:
- Configure Rigid or Relaxed scans over bond lengths, angles, or dihedrals.
- Visualize potential energy surfaces with interactive plots and trajectory animations.
- Transition State Analysis:
- Perform Transition State Optimizations (requires
geomeTRIClibrary). - Visualize imaginary frequencies (saddle points) with animated vibrational modes.
- Perform Transition State Optimizations (requires
Robust Job Management
- Organized Output: Each calculation automatically creates a unique directory (output/job_1, job_2…) to prevent data loss.
- Full Logging: Captures all PySCF output (including low-level C warnings) to both the GUI log window and pyscf.out log files.
- Artifact Safety: Inputs, Checkpoints, and Cube files are strictly contained within their specific job folder.
Installation
Requirements
- PySCF
- PyQt6
- NumPy
- GeomeTRIC
- Matplotlib
pip install pyscf PyQt6 numpy geometric matplotlib
[!WARNING] PySCF installation may fail on Windows, so it may only work on MacOS or Linux.
Setup
- Download: Download the plugin from the Plugin Explorer.
- Install: Unzip the downloaded file and place the
pyscf_calculatorfolder into your application’spluginsdirectory. - Launch: Start the main application, then navigate to the Extensions menu and select PySCF Calculator.
Usage
- Setup Tab: Load your molecule (XYZ format), select method/basis, and configure resources (Threads/Memory).
- Run: Click “Run Calculation”. The interface will switch to the Visualization tab upon completion.
- Visualize:
- Use the Orbital Diagram to inspect electronic structure.
- Select specific orbitals (e.g., HOMO, LUMO) to generate Cube files.
- Click “Show Properties” for thermodynamic data (after Frequency jobs).
License
This project is licensed under the GNU General Public License v3.0 (GPLv3). See the LICENSE file for details.