MoleditPy ユーザーマニュアル
目次
- 1. はじめに
- 2. インストールと起動
- 3. 画面構成
- 4. 基本的な操作 (2D 編集ビュー)
- 5. 3D 機能
- 6. ファイル操作
- 7. 分子分析
- 8. 設定
- 9. プラグインシステム
- 10. キーボードショートカット
- 11. バージョン情報・ライセンス
1. はじめに
MoleditPy は、Python で開発された分子編集ソフトウェアです。直感的なインターフェースを通じて、2D での分子構造の描画・編集、および 3D 構造の生成・表示・編集を行うことができます。
主な機能:
- 2D 分子編集: 原子や結合の追加・削除・変更、テンプレートを用いた構造作成。ユーザーが指定した構造を保存・利用できるユーザーテンプレートにも対応しています。
- 3D 構造生成: RDKit や Open Babel (オプション) を利用した 2D 構造からの 3D 座標生成および構造最適化。
- 3D 分子表示: Ball & Stick, CPK (空間充填), Wireframe, Stick など、複数のスタイルでの 3D 分子表示。
- ファイル操作: 独自形式 (.pmeprj) でのプロジェクト保存・読み込み、標準的な化学ファイル形式 (MOL, SDF, XYZ) や SMILES, InChI 文字列のインポート・エクスポートに対応。エクスポートされたファイルは、DFT計算へ使用することができます。画像 (PNG) や 3D印刷用の3D モデル (STL, OBJ) としてのエクスポートも可能。
- 分子分析: SMILES, InChI, 分子式, 分子量, LogP, TPSA などの基本的な分子特性を表示。
- 3D 測定: 3D ビュー上で原子間の距離、3 原子間の角度、4 原子間の二面角を測定。
- 3D 編集: 分子全体の平行移動、選択原子の平面化、特定軸への整列、結合長・角度・二面角の調整、鏡像作成。
- カスタマイズ: 3D 表示の背景色、ライティング、表示スタイル詳細などを設定可能。
![]()
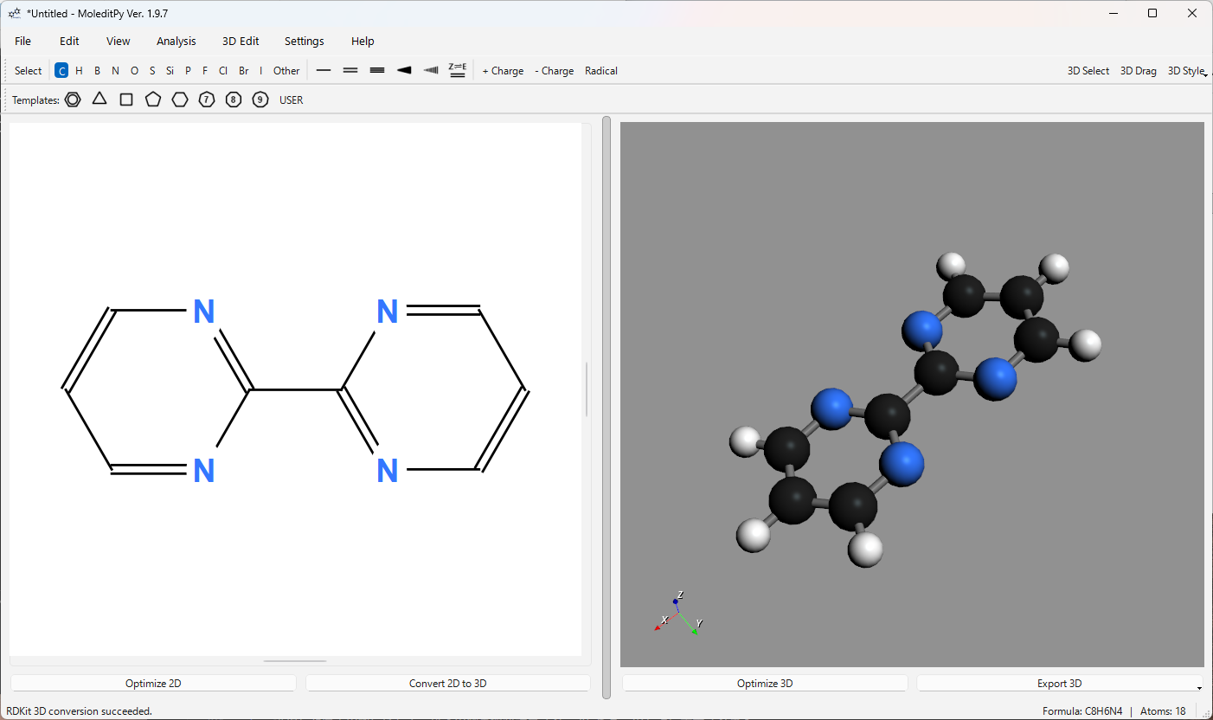
2. インストールと起動
-
パッケージのインストール このコマンドを実行すると、お使いのOS(Windows/macOSまたはLinux)に適した
moleditpy本体が自動的にインストールされます。pip install moleditpy-installer -
ショートカットの作成 インストール後、このコマンドを実行すると、アプリケーションメニュー(スタートメニューやアプリケーションフォルダなど)にショートカットが作成されます。
python -m moleditpy_installer
起動:
以下のコマンドで起動できます。
moleditpy
コマンドラインオプション:
| オプション | 説明 |
|---|---|
moleditpy [ファイル] |
起動時に指定したファイルを開く |
moleditpy --version |
バージョン番号を表示して終了する |
moleditpy --safe |
セーフモードで起動する: 起動時にすべてのプラグインの読み込みをスキップします。プラグインが原因でクラッシュしたり、起動できない場合にお使いください。 |
moleditpy --install-plugin [パス] |
プラグイン (.py, .zip, フォルダ) をヘッドレスモードでインストールします(要手動確認)。 |
Windows向けインストーラー
Moleditpy は Windows 用のインストーラーを提供しています。インストーラーを使うことで、Python 環境の手動セットアップなしに簡単に Moleditpy をインストールできます。
3. 画面構成
MoleditPy のメインウィンドウは、主に以下の要素で構成されています。
- メニューバー: ファイル操作、編集、表示、分析、設定、ヘルプなどの機能が含まれます。
- メインツールバー: 原子・結合の描画モード選択、電荷・ラジカル設定、3D 表示スタイル変更などの基本的なツールが配置されています。
- テンプレートツールバー: 環状構造などのテンプレートを選択するためのボタンが配置されています。
- 2D 編集ビュー: 分子構造を描画・編集するメインのキャンバスです。マウス操作で原子や結合を追加・編集できます。
- 3D 表示ビュー: 生成または読み込んだ分子の 3D 構造を表示します。マウスで回転、ズーム、パン操作が可能です。3D 測定や編集もこのビューで行います。
- スプリッター: 2D ビューと 3D ビューの間の境界線です。ドラッグして各ビューの表示領域サイズを変更できます。メニューの
View>Panel Layout(または Ctrl+1, 2, 3) で分割比率を素早く切り替えることも可能です。 - ステータスバー: 現在の操作モード、メッセージ、計算中の分子式・原子数などを表示します。
4. 基本的な操作 (2D 編集ビュー)
2D 編集ビューでは、マウスとツールバー、キーボードショートカットを使って分子構造を作成・編集します。
4.1. 描画モードの選択
メインツールバーのボタンをクリックするか、対応するキーボードショートカットを押して、描画モードを選択します。
- Select (Space): 原子や結合を選択、移動するモード。
- 原子ボタン (C, H, N, O など): 対応する元素の原子を追加するモード。クリックで原子を配置、ドラッグで原子と結合を追加。
- 結合ボタン (単結合, 二重結合など): 対応する種類の結合を追加・変更するモード。原子間をドラッグして結合を作成、既存の結合をクリックして種類を変更。
- 電荷ボタン (+/-): 原子をクリックして電荷を増減させるモード。右クリックで電荷を 0 にリセット。
- ラジカルボタン (Radical): 原子をクリックしてラジカル電子数をトグル (0 -> 1 -> 2 -> 0) するモード。右クリックで 0 にリセット。
- テンプレートボタン (ベンゼン環など): キャンバスをクリックしてテンプレート構造を追加するモード。既存の原子や結合にスナップして融合させることも可能。
4.2. 原子・結合の操作
- 原子の追加:
- ツールバーで追加したい元素ボタンを選択 (例: ‘C’)。
- キャンバスの任意の位置をクリックすると、その元素の原子が配置されます。
- 結合の追加:
- ツールバーで追加したい結合ボタンを選択 (例: 単結合)。
- 原子 A の上でマウスボタンを押し、原子 B までドラッグして離すと、A と B の間に結合が作成されます。
- 空白領域でドラッグを開始し、原子 A 上で離すと、空白領域の開始点に新しい原子 (デフォルトは炭素) が作られ、原子 A との間に結合が作成されます。
- 原子 A 上でドラッグを開始し、空白領域で離すと、離した位置に新しい原子 (デフォルトは炭素) が作られ、原子 A との間に結合が作成されます。
- 原子・結合の選択:
- ツールバーで ‘Select’ モードを選択。
- 原子または結合をクリックして選択 (複数選択は Shift + クリック)。
- キャンバスの空白領域をドラッグして矩形選択。
- 移動:
- ‘Select’ モードで、選択した原子または結合をドラッグします。原子を移動すると、接続している結合も追従します。
- 削除:
- ‘Select’ モードまたは任意の描画モード (テンプレート、電荷、ラジカル以外) で、削除したい原子または結合を右クリックします。
- ‘Select’ モードでアイテムを選択し、
DeleteまたはBackspaceキーを押します。
- 元素の変更:
- ツールバーで変更したい先の元素ボタンを選択。
- 既存の原子をクリックすると、その原子の元素が変更されます。
- または、マウスカーソルを原子上、ホバー状態で、キーボードショートカット (例: ‘N’ で窒素) を押します。
- 結合次数の変更:
- ツールバーで変更したい先の結合ボタンを選択。
- 既存の結合をクリックすると、その結合の種類が変更されます。
- または、’Select’ モードで結合を選択し、キーボードショートカット (例: ‘2’ で二重結合) を押します。
- 立体結合 (Wedge/Dash) の設定:
- ツールバーで Wedge (太線) または Dash (破線) ボタンを選択。
- 既存の単結合をクリックすると、立体表示が設定されます。もう一度クリックすると方向が反転します。
- または、’Select’ モードで単結合を選択し、’W’ (Wedge) または ‘D’ (Dash) キーを押します。
- 二重結合の E/Z 配置:
- ツールバーで ‘Toggle E/Z’ ボタン (Z⇌E アイコン) を選択。
- 既存の二重結合をクリックすると、立体配置が Z -> E -> (指定なし) の順にトグルします。
- または、結合にマウスカーソルを合わせ、’Z’ または ‘E’ キーを押します。
4.3. テンプレートの使用
- 標準テンプレート: テンプレートツールバーのボタン (ベンゼン環、シクロヘキサン環など) をクリックしてモードを選択し、キャンバスをクリックして配置します。既存の原子や結合の上でクリックすると、構造を融合できます。テンプレートのスナップ距離やフュージング設定は
Settingsメニューから調整可能です。テンプレートの配置中にAltキーを押し続けると、一時的に原子融合(他の頂点が近隣の原子と結合する動作)を無効にして配置することができます。また、Altキーを押していない間は、融合先となる近接原子に合わせてテンプレートのプレビュー形状が動的に変形し、融合後の形状を視覚的にプレビュー表示します。 - ユーザーテンプレート:
- テンプレートツールバーの ‘USER’ ボタンをクリックするか、メニューの
File>Save 2D as Template...で現在の構造をテンプレートとして保存します。 - ‘USER’ ボタンをクリックすると、ユーザーテンプレートダイアログが開きます。
- ダイアログで使いたいテンプレートをクリックすると、そのテンプレートを使用するモードになります。キャンバスをクリックして配置します。ダイアログは開いたままなので、連続して異なるテンプレートを使用できます。
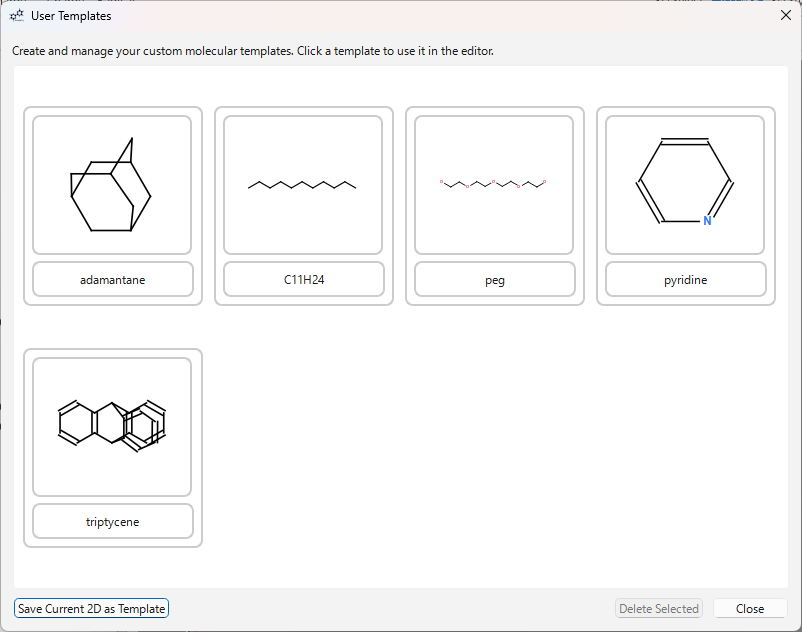
- テンプレートツールバーの ‘USER’ ボタンをクリックするか、メニューの
4.4. Undo/Redo
- メニューの
Edit>Undo(Ctrl+Z) およびRedo(Ctrl+Y / Ctrl+Shift+Z) を使用して、操作を取り消したり、やり直したりできます。
4.5. コピー/カット/ペースト
- ‘Select’ モードで原子や結合を選択し、メニューの
Edit>Copy(Ctrl+C) またはCut(Ctrl+X) を実行します。 - メニューの
Edit>Paste(Ctrl+V) を実行すると、クリップボードの内容がカーソル位置に貼り付けられます。MoleditPy 内部のコピー&ペーストに対応しています。
4.6. その他の編集・表示機能
- Clean Up 2D: 2D 構造をきれいに整列させたい場合は、左下の
Clean Up 2Dボタンをクリックするか、キーボードのCtrl+Jを押します。 - Rotate 2D…: メニューの
Edit>Rotate 2D...(Ctrl+R) を選択すると、角度を指定して 2D 分子を回転させるダイアログが開きます。ショートカットキーとしても利用できます。 - Add Hydrogens: メニューの
Edit>Add Hydrogensを選択すると、現在の結合状態に基づいて水素原子を明示的に追加します。 - Remove Hydrogens: メニューの
Edit>Remove Hydrogensを選択すると、すべての水素原子を削除します。 - Clear All: キャンバス上のすべての原子と結合を削除するには、メニューの
Edit>Clear All(Ctrl+Shift+C) を実行します。 - Show Chiral Labels: メニューの
View>Show Chiral Labelsをチェックすると、不斉中心の R/S ラベルを表示できます。
5. 3D 機能
MoleditPy は、描画した 2D 構造から 3D 構造を生成し、表示、測定、編集を行う機能を提供します。
5.1. 2D から 3D への変換
- 2D 編集ビューで分子構造を描画します。
- 左下の Convert 2D to 3D ボタンをクリックするか、メニューの
Edit>Convert 2D to 3D(Ctrl+K) を選択します。 - 計算が開始され、ステータスバーに進捗が表示されます。RDKit (ETKDGv2 アルゴリズム) を使用して 3D 座標が生成され、力場計算 (MMFF94 または UFF) によって簡単な構造最適化が行われます。
- 成功すると、3D 表示ビューに生成された 3D 構造が表示されます。
(設定): メニューの Settings > 3D Conversion から、変換に使用するライブラリ (RDKit, Open Babel) の優先順位を設定できます。また、Direct モード(2D 座標をそのまま用い、水素原子を付加するモード)を選択することもできます。Open Babel がインストールされていない場合、関連するオプションは無効になります。
変換モードの詳細:
- Fallback モード(デフォルト): RDKit $\rightarrow$ Open Babel(利用可能な場合) $\rightarrow$ Direct モードの順に自動で実行を試みます。
- RDKit モード: RDKit の ETKDGv2 (Experimental-Torsion Knowledge Distance Geometry) アルゴリズムを用いてコンフォーマーを生成します。生成に失敗した場合は、立体化学の制約を明示的に加えた距離限界マトリックスによるトライアングル・スムージングを適用し、再度埋め込みを試みます。
- Open Babel モード: Open Babel の
make3D()座標ジェネレーターを使用します。プログラムのハングアップやクラッシュを防ぐため、バックグラウンドの分離されたサブプロセスで安全に実行されます。 - Direct モード: 3Dコンフォーマー生成を行わず、2Dキャンバスの配置をそのまま保持します。重原子は $Z = 0.0$ 平面に配置され、不足している水素原子が幾何学的に追加されます。立体化学は、Wedge/Dash結合を持つ原子にZ座標のオフセット ($Z = \pm 1.5$ Å) を加えることで再現されます。
5.2. 3D 構造の最適化
- 3D 構造が表示されている状態で、右下の Optimize 3D ボタンをクリックするか、メニューの
Edit>Optimize 3D(Ctrl+L) を選択します。 - 選択されている力場 (MMFF または UFF) を用いて、より詳細な構造最適化計算が実行されます。
- 最適化手法が失敗した場合、一時的なUFFオーバーライドを提案するインタラクティブなフォールバックプロンプトが表示されることがあります。
- 完了すると、最適化された構造が 3D ビューに再描画されます。
(設定): メニューの Settings > 3D Optimization Settings から、使用する力場計算ライブラリとメソッド (RDKit MMFF94/MMFF94s/UFF, Open Babel MMFF94/MMFF94s/UFF/GAFF/Ghemical) を選択できます。
(クイック選択): Optimize 3D ボタンを右クリックすると、デフォルトの選択を変更せずに、任意のメソッドで一度だけ最適化を実行できます。
構造最適化メソッドの詳細:
- RDKit バックエンド: MMFF94s (分光学的/デフォルト), MMFF94, および UFF (Universal Force Field) をサポートします。遷移金属などの未サポート原子が含まれることで MMFF94/MMFF94s のセットアップが失敗した場合、クラッシュを防ぐために自動的に UFF にフォールバックします。
- Open Babel バックエンド: MMFF94s, MMFF94, UFF, GAFF (General Amber Force Field), および Ghemical をサポートします。原子間の重なりや衝突を解消するため、最急降下法 (Steepest Descent) を100反復実行した後に、共役勾配法 (Conjugate Gradients) でエネルギー最小化を実行します。
- プラグインメソッド: インストールされたプラグインは独自の最適化手法を追加できます。存在する場合、組み込みの力場と並んで
Settings>3D Optimization Settings(デフォルトとして選択可能)および Optimize 3D の右クリックメニューに表示されます。
5.3. 3D 表示スタイルの変更
メインツールバー右側の 3D Style ドロップダウンメニューから、表示スタイルを選択できます。
- Ball & Stick: 原子を球 (ファンデルワールス半径の縮小版)、結合を棒で表示します。標準的なスタイルです。
- CPK (Space-filling): 原子をファンデルワールス半径に基づいた空間充填球で表示します。分子の体積や形状を視覚化するのに適しています。
- Wireframe: 結合のみを細い線で表示します。原子は表示されません。
- Stick: 結合を太い棒で、原子を小さな球で表示します。
- Aromatic Ring (芳香環): デフォルトでは、芳香環 (ベンゼン環など) は単結合で表示されます。設定により芳香環 (トーラス) やケクレ構造 (二重結合の交互配置) に変更することができます。
(設定): 各表示スタイルの詳細 (原子サイズ、結合半径、描画品質、多重結合の表示オフセットと太さなど) は、メニューの Settings > Settings... から調整できます。また、Settings > CPK Colors... から CPK色 を変更することも可能です。
5.4. 3D ビューの操作
- 回転: マウスの左ボタンドラッグ。
- ズーム: マウスホイールの回転 (Ctrl + ホイールでも可)。
- パン (移動): マウスの中ボタンドラッグ、または Shift + 左ボタンドラッグ。
- ビューのリセット: メニューの
View>Reset 3D View(Ctrl+Shift+R) で、カメラの位置とズームを初期状態に戻します。 - 3D 分子の再描画: メニューの
View>Redraw 3D Moleculeで、3D構造の再描画を強制します。
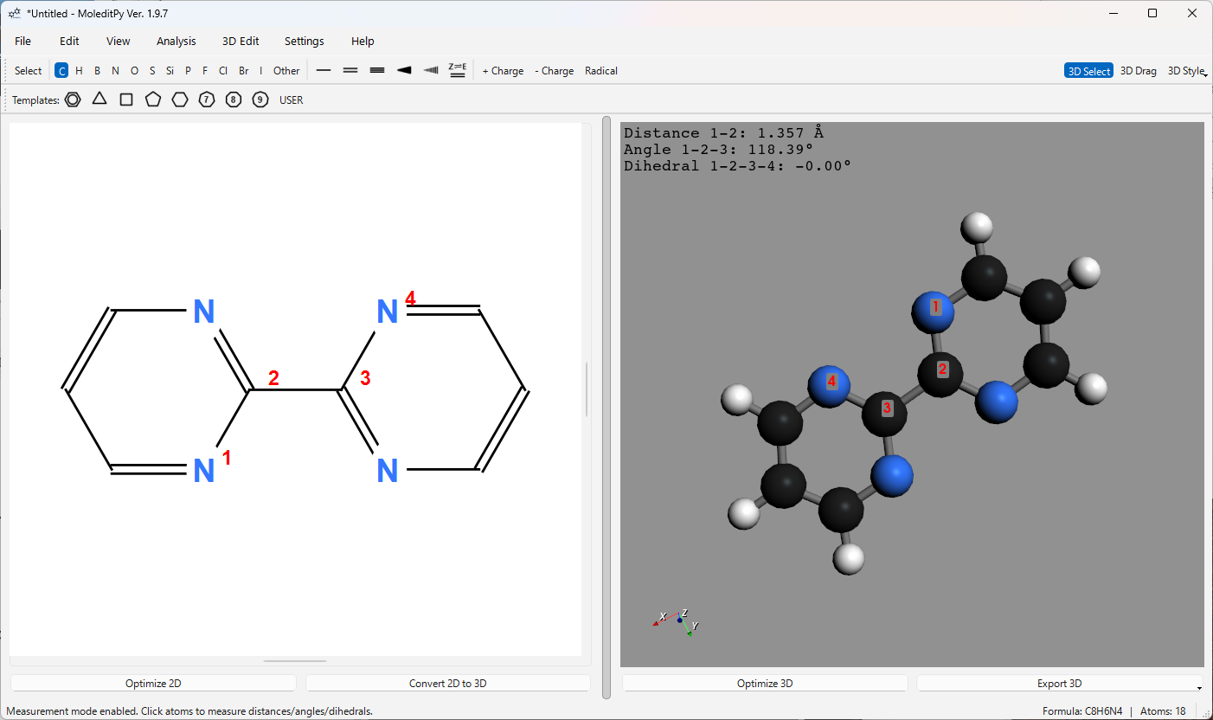
5.5. 3D 測定機能 (“3D Select” モード)
- メインツールバーの 3D Select ボタンをクリックして、測定モードを有効にします。
- 3D ビュー上で原子をクリックして選択します。選択された原子には順番に番号 (1, 2, 3, 4) が赤いラベルで表示されます。
- 選択した原子の数に応じて、以下の測定値が計算され、3D ビューの左上に表示されます。
- 2 原子選択: 原子間距離 (Å)
- 3 原子選択: 距離 (1-2) および角度 (1-2-3) (°)
- 4 原子選択: 距離 (1-2)、角度 (1-2-3) および二面角 (1-2-3-4) (°)
- 原子以外をクリックするか、または 3D Select ボタンを再度クリックしてモードを解除すると、選択と測定値がクリアされます。
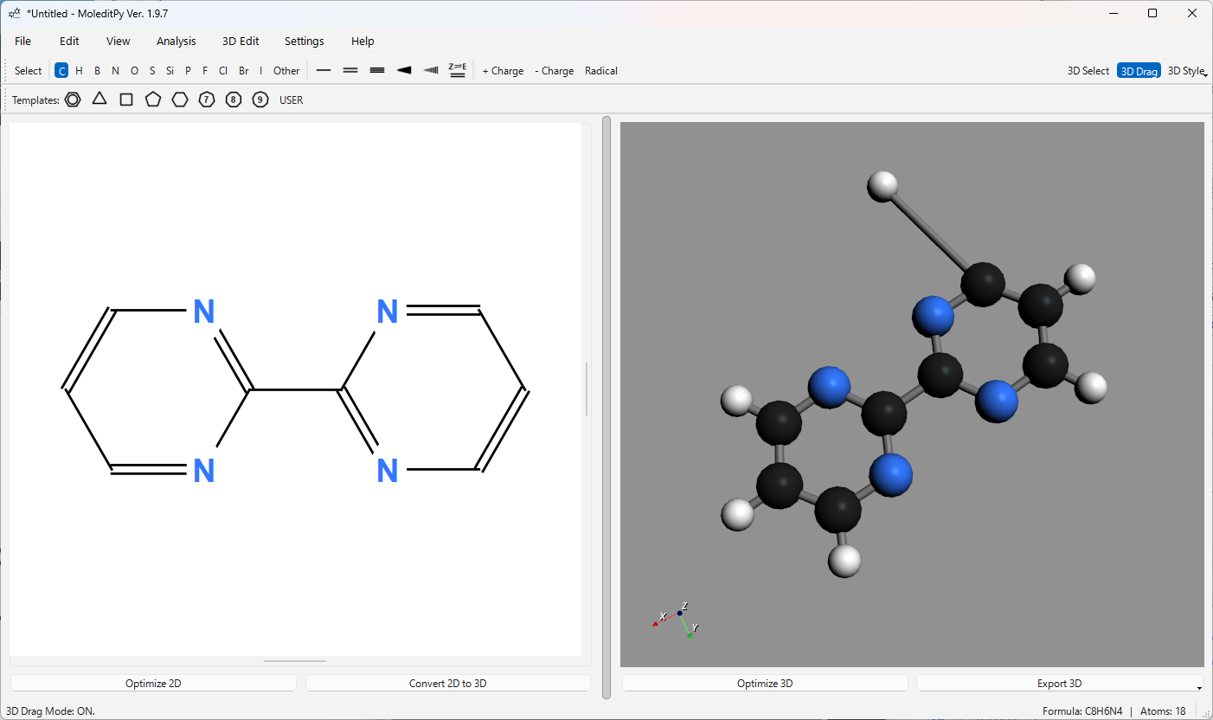
5.6. 3D 編集機能 (“3D Drag” モード / Alt キー)
3D 構造の原子座標を直接編集できます。メインツールバーの 3D Drag ボタンをオンにするか、Alt キーを押しながら操作します。
- 原子のドラッグ: 3D Drag モード中、原子をクリックしてドラッグすると、その原子を 3D 空間内で移動できます。マウスボタンを離すと位置が確定します。
5.7. その他の 3D 編集機能 (メニュー 3D Edit)
これらの機能は、3D 構造が表示されている場合にメニューから利用できます。多くは専用のダイアログが開き、そこで原子を選択したり、パラメータを入力したりします。
- Translation…: 分子全体または選択した原子群を平行移動します。相対座標 (移動量) を指定するか、絶対座標モードを使用して選択範囲を 3D 空間の正確な位置に移動させることができます。選択した原子のみを平行移動するオプションもあります。
- Move Selected Atoms…: 選択された原子のみを平行移動または回転させます。ダイアログから選択原子の重心を中心とした正確な平行移動量(dX, dY, dZ in Å)や回転角度(X/Y/Z軸まわりの角度)を入力できるほか、3Dビュー上で選択原子を直接ドラッグ操作(左ドラッグで平行移動、右ドラッグで回転)して編集することも可能です。
-
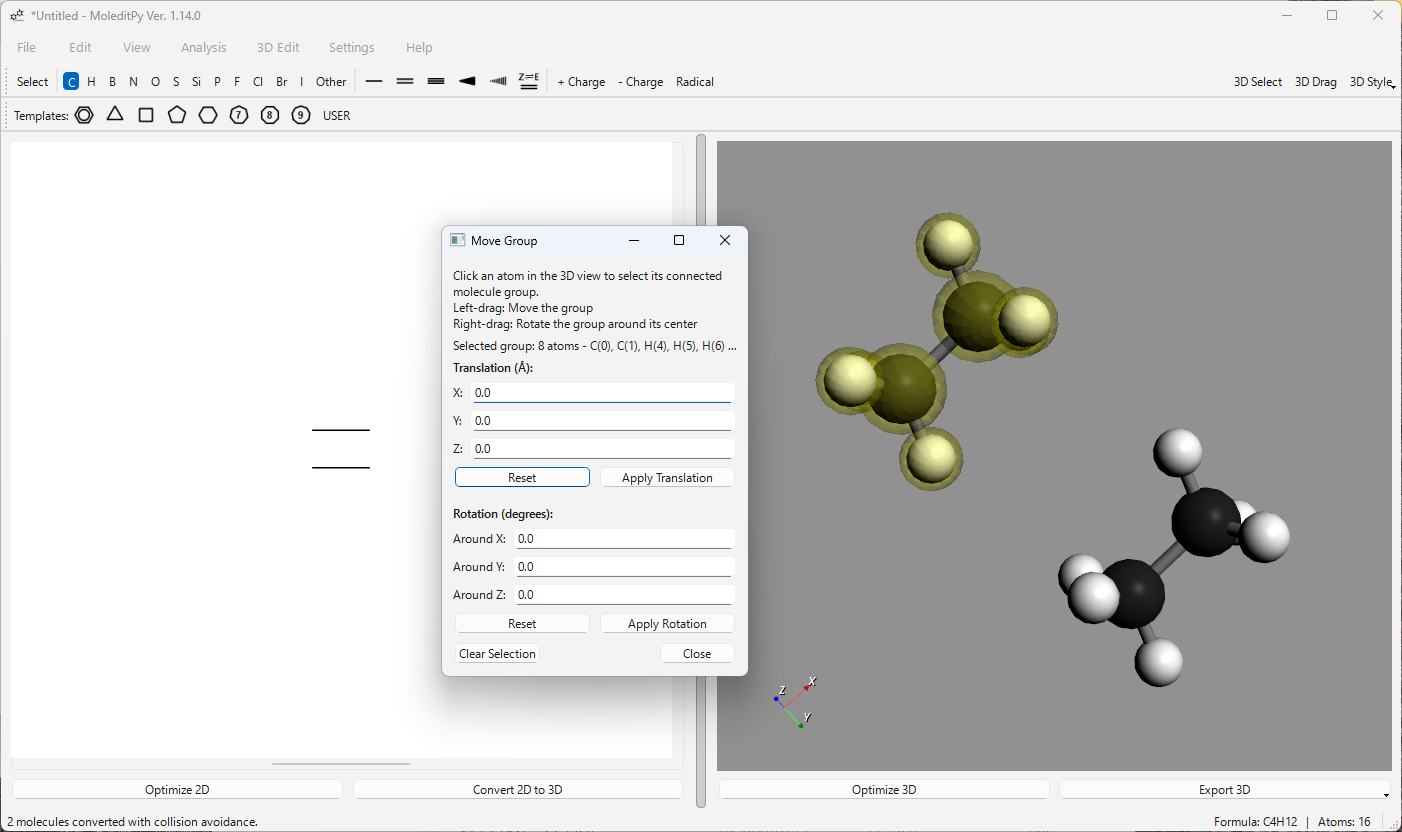
Move Group…: 接続された分子フラグメント(グループ)の選択と操作を可能にします。
- 選択: 3Dビューで、原子を左クリックすると、接続されたグループ全体が選択されます(黄色でハイライト表示)。Ctrl + 左クリックで、選択にグループを追加/削除できます。
- インタラクティブな移動: ハイライト表示された原子を左ドラッグすると、選択されたすべてのグループが移動します。
- インタラクティブな回転: ハイライト表示された原子を右ドラッグすると、選択されたすべてのグループが結合重心を中心に回転します。
- 数値入力: ダイアログを使用して、選択されたすべてのグループに正確な平行移動(Å)または回転(度)を適用します。
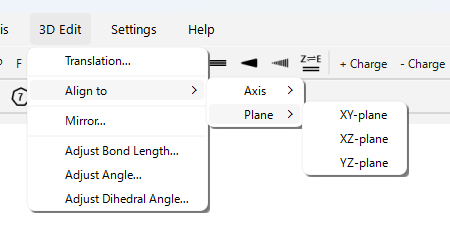
- Align to > Axis > (X/Y/Z)-axis…: 選択した 2 原子を結ぶ線が、指定した座標軸 (X, Y, または Z) に沿うように、分子全体を回転・移動します (1 番目の原子が原点に、2 番目の原子が軸上に配置されます)。
- Align to > Plane > (XY/XZ/YZ)-plane…: 選択した 3 原子以上を含む平面が、指定した座標平面 (XY, XZ, または YZ) と平行になるように、分子全体を回転します。
- Mirror…: 分子全体の鏡像を指定した平面 (XY, XZ, YZ) に対して作成します。
- Adjust Bond Length…: 選択した 2 原子間の距離を指定した値に変更します。片方の原子(または接続グループ)を固定するか、両方を動かすか選択できます。リアルタイム調整用のインタラクティブなスライダーも備えています。
- Adjust Angle…: 選択した 3 原子 (1-2-3) がなす角度を指定した値に変更します。原子 3 側(または接続グループ)を回転させるか、両腕を均等に回転させるか選択できます。リアルタイム調整用のインタラクティブなスライダーも備えています。
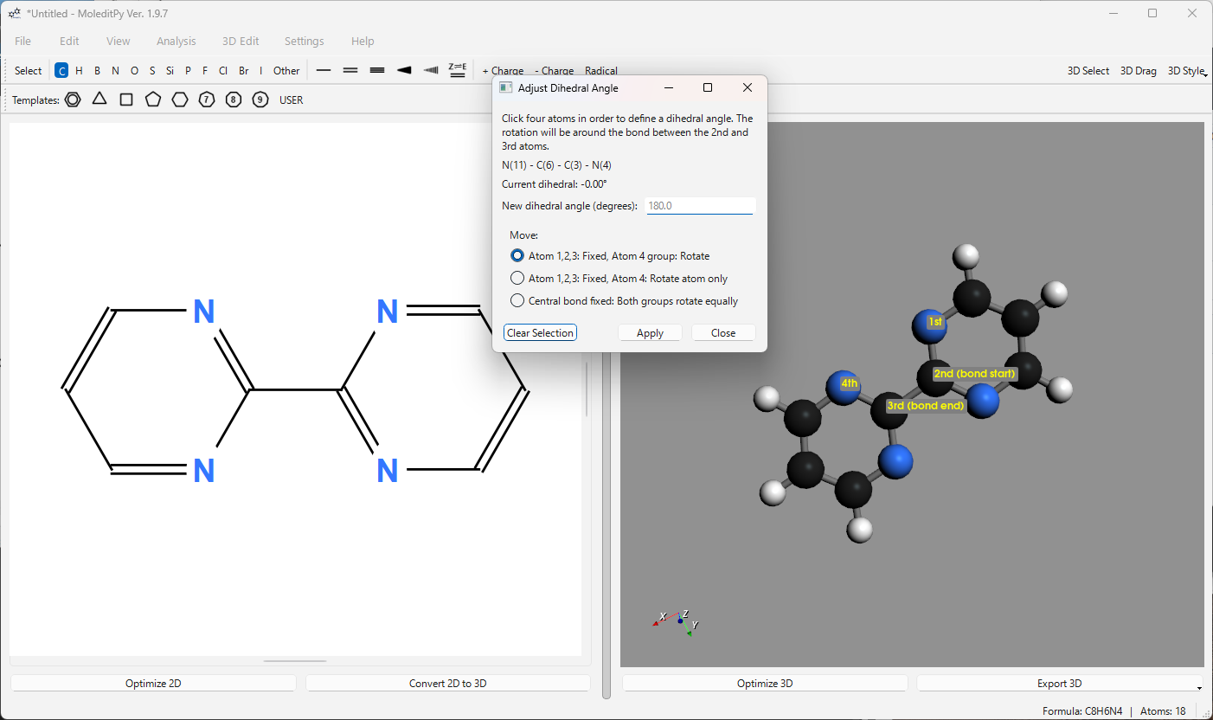
- Adjust Dihedral Angle…: 選択した 4 原子 (1-2-3-4) がなす二面角を指定した値に変更します。原子 4 側(または接続グループ)を回転させるか、両グループを均等に回転させるか選択できます。リアルタイム調整用のインタラクティブなスライダーも備えています。
- Planarize…: 選択した 3 個以上の原子について最小二乗法で最適平面を計算し、それらの原子をその平面に射影(平面化)します。
5.8. 制約付き最適化 (Constrained Optimization)
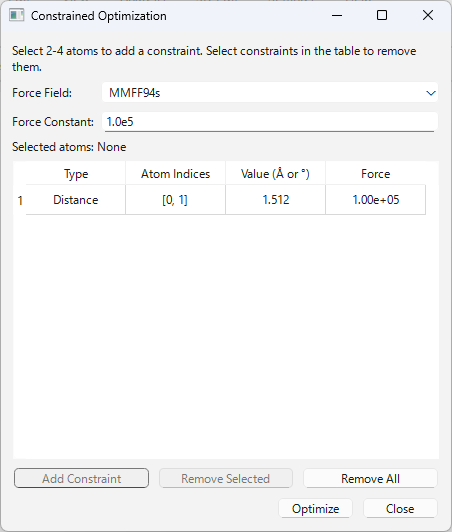
特定の原子間の距離、角度、または二面角の値を固定(制約)したまま、分子全体の構造最適化(力場計算)を実行する高度な機能です。
- 制約の追加:
3D Edit>Constrained Optimization...を選択し、ダイアログを開きます。- 3Dビューで原子を 2 個(距離)、3 個(角度)、または 4 個(二面角)クリックして選択します。選択した原子は黄色いラベル (A1, A2…) でハイライトされます。
- 「Add Constraint」ボタン(例: “Add Distance Constraint”)を押すと、現在の構造での値がテーブルに追加されます。
- 制約の編集と削除:
- 編集: テーブルの「Value」列をダブルクリック(または選択してEnter)すると、制約値を直接編集できます。
- 削除: テーブルで削除したい制約の行を選択し、「Remove Selected」ボタンを押すか、キーボードの
Delete/Backspaceキーを押します。 - 確認: テーブルの行を選択すると、3Dビューに対応する原子が水色でハイライトされます。
- 最適化の実行:
- 「Force Field」ドロップダウンから使用する力場(MMFF94s, MMFF94, UFF)を選択します。(デフォルトは
Settingsメニューで設定された値が読み込まれます) - 「Optimize」ボタン(または
Enterキー)を押すと、テーブル内のすべての制約を保持したまま最適化が実行されます。
- 「Force Field」ドロップダウンから使用する力場(MMFF94s, MMFF94, UFF)を選択します。(デフォルトは
5.9. 原子情報の表示
メニューの View > 3D Atom Info Display から、3D ビューの各原子の上に表示する情報を選択できます。
- Show Original ID / Index (または Show XYZ Unique ID): 2D エディタ由来の場合は元の ID、XYZ ファイル由来の場合は XYZ ファイル内でのインデックスを表示します。開始インデックスを切り替える 0/1 ベースのサブメニューも含まれています。
- Show RDKit Index: RDKit 内部での原子インデックスを表示します。
- Show Coordinates (X,Y,Z): 各原子の 3D 座標を表示します。
- Show Element Symbol: 各原子の元素記号を表示します。
同じメニュー項目を再度選択すると、表示がオフになります。
6. ファイル操作
メニューバーの File メニューから各種ファイル操作を行います。
6.1. プロジェクトファイル (.pmeprj (Python Molecular Editor Project File))
- New (Ctrl+N): 現在の作業内容をすべてクリアし、新規状態にします。未保存の変更がある場合は確認ダイアログが表示されます。
- Open Project… (Ctrl+O): 以前に保存したプロジェクトファイル (.pmeprj または .pmeraw) を開きます。
- Save Project (Ctrl+S): 現在の作業内容 (2D 構造、生成済みの 3D 構造など) を現在のプロジェクトファイルに上書き保存します。ファイル名が未設定の場合は「名前を付けて保存」ダイアログが開きます。 .pmeprj (JSON 形式) が推奨される形式です。
- Save Project As… (Ctrl+Shift+S): 現在の作業内容を新しい名前または場所でプロジェクトファイル (.pmeprj) として保存します。
6.2. インポート
- Import > MOL/SDF File…: MOL または SDF ファイルを読み込み、2D 構造として表示します。ファイルに 3D 座標が含まれていても、2D 座標が再計算されます (立体化学は保持されます)。インポートされた分子は、既存の構造を上書きすることなく、自動的に現在のキャンバスに追記 (アペンド) されます。
- Import > SMILES…: SMILES 文字列を入力するダイアログを開き、入力された分子を 2D 構造として表示します。インポートされた分子は自動的に現在のキャンバスに追記されます。
- Import > InChI…: InChI 文字列を入力するダイアログを開き、入力された分子を 2D 構造として表示します。インポートされた分子は自動的に現在のキャンバスに追記されます。
- Import > 3D MOL/SDF (3D View Only)…: 3D 座標を持つ MOL/SDF ファイルを読み込み、3D ビューのみに表示します (2D エディタはクリアされます)。3D ビューアモードになります。
- Import > 3D XYZ (3D View Only)…: XYZ ファイルを読み込み、3D ビューのみに表示します (2D エディタはクリアされます)。原子間距離に基づいて結合が推定されます。3D ビューアモードになります。
6.3. エクスポート
- Export > PME Raw Format…: プロジェクトデータを旧式のバイナリ形式 (.pmeraw) で保存します。
- Export > 2D Formats > MOL File…: 現在の 2D 構造を MOL ファイルとして保存します。
- Export > 2D Formats > PNG Image…: 現在の 2D 編集ビューの内容を PNG 画像ファイルとして保存します。背景を透過させるか選択できます。
- Export > 2D Formats > SVG Image…: 現在の 2D 編集ビューの内容を SVG ベクター画像ファイルとして保存します。背景を透過させるか選択できます。
- Export > 3D Formats > MOL File…: 現在表示されている 3D 構造を 3D 座標を持つ MOL ファイルとして保存します。
- Export > 3D Formats > XYZ File…: 現在表示されている 3D 構造を XYZ ファイルとして保存します。
- Export > 3D Formats > PNG Image…: 現在の 3D 表示ビューの内容を PNG 画像ファイルとして保存します。背景を透過させるか選択できます。
- Export > 3D Formats > STL File…: 現在の 3D モデルを STL ファイル (色なし、3D プリント用など) として保存します。
- Export > 3D Formats > OBJ/MTL (with colors)…: 現在の 3D モデルを OBJ ファイルと MTL ファイル (色情報付き) として保存します。
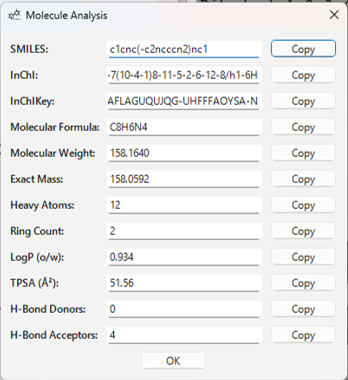
7. 分子分析
メニューの Analysis > Show Analysis... を選択すると、現在 3D ビューに表示されている分子 (XYZ 由来を除く) の基本的な特性を計算し、表示するダイアログが開きます。
表示される情報例:
- SMILES 文字列
- InChI 文字列 / InChIKey
- 分子式
- 分子量
- 精密質量
- 重原子数
- 環の数
- LogP (オクタノール/水分配係数)
- TPSA (極性表面積)
- 水素結合供与体/受容体の数
各値の横にある Copy ボタンで、その値をクリップボードにコピーできます。
8. 設定
メニューの Settings > Settings... から、様々な設定を変更できます。
設定可能な項目:
- 2D Settings タブ:
- View Appearance: 2D 編集ビューの背景色
- Bond Settings:
- 結合色 (Bond Color)
- 結合の太さ (Bond Width)
- 二重・三重結合の間隔 (Spacing)
- 結合端の形状 (Cap Style: Round/Flat/Square)
- 立体結合の太さ (Wedge Bond Width)
- ダッシュ結合の線の数 (Dash Count)
- Atom Settings:
- 原子ラベルのフォントサイズ
- 原子の色を結合色と統一するか (Use Bond Color for Atoms)
- Template Settings:
- テンプレートのスナップ距離 (Snapping Distance): 既存の原子や結合にテンプレートをスナップさせる距離の閾値を設定します。
- 原子の融合 (Enable Atom Fusing): 有効にすると、テンプレートを既存の原子の近くに配置した際に融合 (接続) させます。
- 原子の融合距離 (Fusing Distance): テンプレートの原子が既存の原子と融合する距離の閾値を設定します。
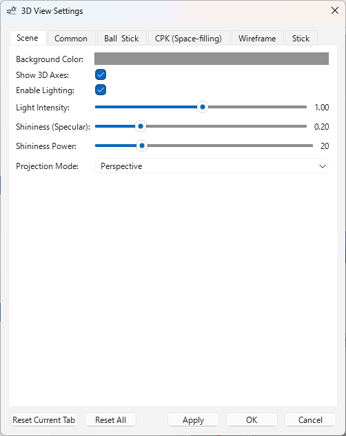
- Scene タブ:
- 背景色
- 3D 座標軸の表示/非表示
- ライティングの有効/無効
- 光の強度
- 表面の光沢 (Specular) とその強さ (Specular Power)
- カメラの投影モード (Perspective / Orthographic)
- 各表示スタイル (Ball & Stick, CPK, Wireframe, Stick) タブ:
- 原子サイズ/半径のスケール
- 結合半径
- 描画品質 (Resolution)
- 多重結合のオプション (オフセット、太さ) (スタイルごと)
- Other タブ:
- XYZ ファイルインポート時に化学的妥当性チェックをスキップするかどうか
- 芳香族系 (ベンゼンなど) をケクレ構造 (二重結合の交互配置) で表示するかどうか
- 芳香環 (トーラス) で表示するかどうか
- 芳香環トーラスの太さ
Apply ボタンで設定を即座に反映し、OK ボタンで適用してダイアログを閉じます。Reset Current Tab / Reset All で設定をデフォルトに戻すこともできます。設定は次回起動時にも保持されます。
メニューの Settings > Reset All Settings を実行すると、メニューからすべての設定を一括で初期状態にリセットできます。
-
Settings メニューのCPK Colors…から、CPK色の変更ができます。
9. プラグインシステム
MoleditPyは、Pythonスクリプトによる機能の拡張をサポートしています。
9.1. プラグインの管理
メニューの Plugin > Plugin Manager… からプラグインマネージャーを開くことができます。この画面では、インストール済みのプラグインの確認、再読み込み、および削除が可能です。
9.2. プラグインのインストール方法
- ドラッグ&ドロップ: プラグインマネージャーを開き、
.pyまたは.zipファイルをウィンドウ内にドラッグ&ドロップすることで簡単にインストールできます。インストール時に、プラグインマネージャーはファイルの SHA-256 ハッシュ を計算して表示するため、インストールを確定する前に完全性を検証できます。 - CLI (コマンドライン): コマンドラインから moleditpy –install-plugin [PATH] を実行して、ヘッドレスでインストールすることも可能です。
- 手動インストール: カスタムスクリプトを直接 ~/.moleditpy/plugins フォルダに配置することでも追加できます。
9.3. プラグインの検索と例
公式のプラグインは、Plugin Explorer (https://hiroyokoyama.github.io/moleditpy-plugins/explorer/) から検索・ダウンロードできます。
[!TIP] Plugin Installer プラグインをインストールして使用することを強くお勧めします。これにより、MoleditPy 内から直接他のプラグインを簡単に閲覧・インストールできるだけでなく、ダウンロードしたプラグインの SHA-256 ハッシュを公式データベースと自動的に照合し、安全性を検証してくれます。
更新された利用可能なプラグインの例:
- ORCA Input Generator Pro: 複雑なブロック構築を支援するブロックビルダーを備えた、ORCAの入力生成プラグインです。
- PySCF Calculator: PySCFを使用して計算を直接実行できるプラグインです。
- ORCA Result Analyzer: ORCAの計算結果を解析・可視化するためのプラグインです。
10. キーボードショートカット
| キー | 機能 (Select モード) | 機能 (描画/編集モード) |
|---|---|---|
Space |
Select モードに切り替え / 全選択 (選択なしの場合) | Select モードに切り替え |
C, H, N, O, S, P, F, I, B |
対応する原子描画モードに切り替え / カーソル下の原子をその元素に変更 | 対応する原子描画モードに切り替え |
Shift+C |
Cl 描画モード / カーソル下の原子を Cl に変更 | Cl 描画モード |
Shift+S |
Si 描画モード / カーソル下の原子を Si に変更 | Si 描画モード |
Shift+B |
Br 描画モード / カーソル下の原子を Br に変更 | Br 描画モード |
1 |
単結合描画モード / 選択結合 or カーソル下結合を単結合に / 原子に原子を追加 | 単結合描画モード |
2 |
二重結合描画モード / 選択結合 or カーソル下結合を二重結合に | 二重結合描画モード |
3 |
三重結合描画モード / 選択結合 or カーソル下結合を三重結合に | 三重結合描画モード |
4 |
(空の空間で)ベンゼンモードに切り替える / (原子/結合上で)ベンゼンを配置する | ベンゼンテンプレートモード |
W |
Wedge 結合描画モード / 選択結合 or カーソル下結合を Wedge に (クリックで反転) | Wedge 結合描画モード |
D |
Dash 結合描画モード / 選択結合 or カーソル下結合を Dash に (クリックで反転) | Dash 結合描画モード |
Z / E |
(カーソル下の二重結合に対して) Z / E 配置を指定 | (カーソル下の二重結合に対して) Z / E 配置を指定 |
. (ピリオド) |
選択原子 or カーソル下原子のラジカルをトグル (0->1->2->0) | 選択原子 or カーソル下原子のラジカルをトグル (0->1->2->0) |
+ / - |
選択原子 or カーソル下原子の電荷を増減 | 選択原子 or カーソル下原子の電荷を増減 |
Alt |
(3Dビュー) 長押しで原子をドラッグ (3Dドラッグのショートカット) | (2Dビュー) テンプレート配置中に長押しで原子融合を無効化 |
Delete / Backspace |
選択アイテム or カーソル下のアイテムを削除 | (描画操作中に) 操作をキャンセル / 選択アイテム or カーソル下のアイテムを削除 |
| キー | 機能 |
|---|---|
Ctrl+Z |
Undo |
Ctrl+Y / Ctrl+Shift+Z |
Redo |
Ctrl+C |
Copy Selection |
Ctrl+X |
Cut Selection |
Ctrl+V |
Paste |
Ctrl+A |
Select All |
Ctrl+R |
Rotate 2D Molecule |
Ctrl+N |
New |
Ctrl+O |
Open Project… |
Ctrl+S |
Save Project |
Ctrl+Shift+S |
Save Project As… |
Ctrl+J |
Clean Up 2D |
Ctrl+K |
Convert 2D to 3D |
Ctrl+L |
Optimize 3D |
Ctrl++ |
Zoom In (2D View) |
Ctrl+- |
Zoom Out (2D View) |
Ctrl+0 |
Reset Zoom (2D View) |
Ctrl+9 |
Fit to View (2D View) |
Ctrl+Shift+R |
Reset 3D View |
Ctrl+1 |
Panel Layout 50:50 |
Ctrl+2 |
Panel Layout 70:30 (2D Focus) |
Ctrl+3 |
Panel Layout 30:70 (3D Focus) |
Ctrl+H |
Toggle 2D Panel Visibility |
Ctrl+Q |
Quit |
11. バージョン情報・ライセンス
- バージョン: 4.2
- 作者: Hiromichi Yokoyama
- ライセンス: GPL-3.0 license
- リポジトリ: https://github.com/HiroYokoyama/python_molecular_editor
- DOI: 10.5281/zenodo.17268532
メニューの Help > About からバージョン情報を確認できます。